Thermodynamics
From ab initio calculation the total energies and the normal modes of the molecule are determined. These theoretically derived values refer to:
an isolated molecule
with stationary nuclei. Explicit values of the total energy can only be used to calculate reaction energies, or for comparison between isomers, if the same method, basis set etc. was applied to all molecules! From the total energies, derived thermodynamic properties describe a
gas phase system at 0K.
The experimental chemist, however, deals mostly with condensed phases or heterogeneous systems. Ways to include the influence of e.g. solvents, into an ab initio calculation, are very limited. In particular, the solvation energy has to be considered, as well as the electrostatic interaction between molecules and solvent molecules. Of course, solvation can have a large effect on structure and energetics. In most cases, it will not be possible to consider solvation effects. Therefore, the use of standard ab initio results calculated for isolated molecules, for describing a condensed phase system can be done, but much care should be taken in interpreting the results.
Molecular reaction properties (N = number of species involved in the reaction, the reactants) are most interesting from the thermodynamic point of view:
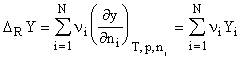
where can be a thermodynamic reaction property such as the internal energy, U, (free) enthalpy, or entropy for a generalized reaction: (R = reactants):
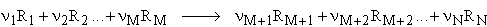
By definition, the stoichiometric coefficients, ni , are accordingly

either negative, for the starting materials, or positive for the products. Applying thermodynamic laws, the corresponding thermodynamic properties can be derived from the total energies at 0 Kelvin.
Chemists are usually interested in the molecular reaction enthalpy, DRH . The relationship between the (internal) molecular reaction energy, DRU, and DRH can be derived from the differentiation of h = u + pv (p = pressure, v = volume), with respect to dni, at constant temperature, T, and pressure, p (work done due to the change in volume is considered):
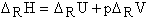
Assuming an ideal gas:

Considering the work done due to the change in volume, the molecular reaction enthalpy can only be calculated by the above equation at 0 Kelvin, i.e. DRU = DRH. At temperatures above 0 K, the temperature corrected molecular reaction energy is needed to estimate the molecular reaction enthalpy.
To calculate the reaction enthalpy at a desired temperature, the following expression has to be used:

Here, T1 = 0 K and T2 is generally 298 K. DRCp represents the molecular reaction heat capacity (at constant pressure):
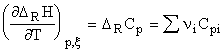
If the exact frequencies of the normal modes are known, the corrections needed are straightforward, by applying statistical thermodynamics for ideal gases within the approximation of the harmonic oscillator and the rigid rotator. Then, the molecular enthalpy is given by:

with
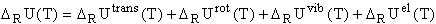
For high temperatures, when quantum mechanical effects are negligible for translation and rotation, the first two terms can be written as:

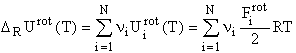
with = 2 for a linear and 3 for a non-linear molecule.
At the absolute zero point, the atoms in the molecules still vibrate around the equilibrium position. Therefore, besides the temperature correction, a zero point vibrational correction (ZPE zero point energy) must be made. According to Heisenberg’s uncertainty principle,
it is not possible to determine absolutely precisely, the position and the momentum of a particle at the same time; thus there must remain some vibrational energy at the absolute zero point, which cannot fall below the zero point energy.
In principle, two "zero points" could be defined: One is the minimum point of the potential curve. The other is the vibrational ground state. The first one is chosen so that -De corresponds to the electronic ground state and D0 corresponds then to the energy difference D0 = De - zpe between the lowest vibrational ground state and the dissociated molecule. The temperature correction can therefore be expressed as:
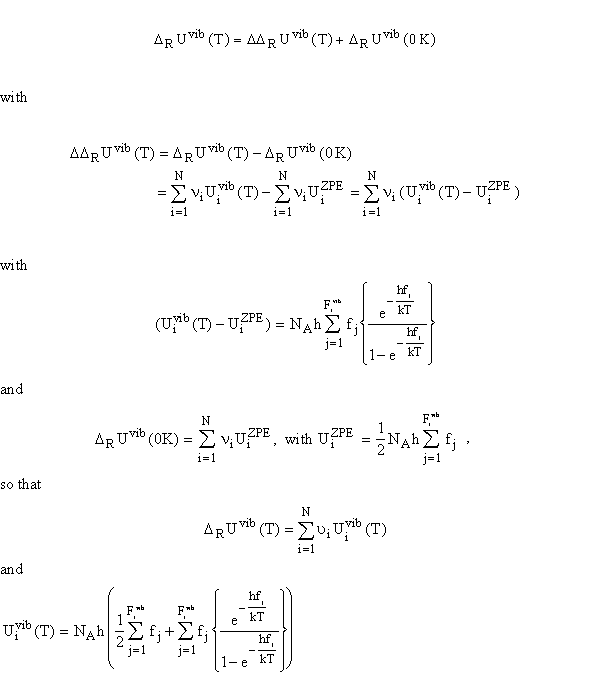
The excitation of the vibrations in a molecule depends on the bond strengths between the atoms and on the masses. At 298 K most vibrations remain unexcited, so that the vibrational correction is mostly a correction to the zero point vibrational energy. At very high temperature, the contribution, DDRUvib(T), converges towards RT for each vibrational degree of freedom. For example, for typical two-atomic gases 2000 to 6000 K are needed to excite all vibrational modes.
If a scaling factor is employed for the calculated frequencies, due to the applied harmonic approximation, then in the statistical correction (as described before), these scaled vibrational frequencies must also be used.
For the electronic term, the approximation that at temperature, T, the molecules are still in the electronic ground state ( i.e. as at O K) can be used:

where Ei(tot,QM) represents the ab initio (or generalized, quantum-mechanically) determined total energy. The contribution of excited electronic states is usually extremely small, as the thermal energy, kT, for any realistic temperature is much too small ( kT<<e) to excite electrons from the ground state (e.g. for a two-atomic gas is ca. e = 10eV).